Nucleophilic Aromatic Substitution
Study Notes
A nucleophilic aromatic substitution reaction is a reaction in which one of the substituents in an aromatic ring is replaced by a nucleophile.
A Meisenheimer complex is a negatively charged intermediate formed by the attack of a nucleophile upon one of the aromatic-ring carbons during the course of a nucleophilic aromatic substitution reaction. A typical Meisenheimer complex is shown in the reaction scheme below. Notice how this particular complex can be formed from two different starting materials by using a different nucleophile in each case.

Figure 16.2: The formation of a typical Meisenheimer complex
A Nucleophilic Aromatic Displacement Reactions of Aryl Halides
The carbon-halogen bonds of aryl halides are like those of alkenyl halides in being much stronger than those of alkyl halides (see Table 4-6). The simple aryl halides generally are resistant to attack by nucleophiles in either SN1 or Sn2 reactions (Table 14-6). However, this low reactivity can be changed dramatically by changes in the reaction conditions and the structure of the aryl halide. In fact, nucleophilic displacement becomes quite rapid
- when the aryl halide is activated by substitution with strongly electron-attracting groups such as N02, and
- when very strongly basic nucleophilic reagents are used.
Addition-Elimination Mechanism of Nucleophilic Substitution of Aryl Halides
Although the simple aryl halides are inert to the usual nucleophilic reagents, considerable activation is produced by strongly electron-attracting substituents provided these are located in either the ortho or para positions, or both. For example, the displacement of chloride ion from 1-chloro-2,4-dinitrobenzene by dimethylamine occurs readily in ethanol solution at room temperature. Under the same conditions chlorobenzene completely fails to react; thus the activating influence of the two nitro groups amounts to a factor of at least 108:
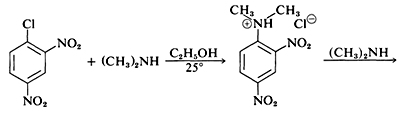
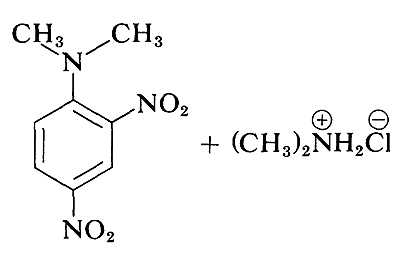
A related reaction is that of 2,4-dinitrofluorobenzene with the amino groups of peptides and proteins, and this reaction provides a means for analysis of the N-terminal amino acids in polypeptide chains. (See Section 25-7B.)
In general, the reactions of activated aryl halides closely resemble the SN2-displacement reactions of aliphatic halides. The same nucleophilic reagents are effective (e.g., CH3O⊖, HO⊖, and RNH2); the reactions are second order overall (first order in halide and first order in nucleophile); and for a given halide the more nucleophilic the attacking reagent, the faster the reaction. However, there must be more than a subtle difference in mechanism because an aryl halide is unable, to pass through the same type of transition state as an alkyl halide in SN2 displacements.
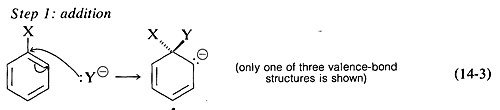
The generally accepted mechanism of nucleophilic aromatic substitution of aryl halides carrying activating groups involves two steps that are closely analogous to those briefly described in Section 14-4 for alkenyl and alkynyl halides. The first step involves attack of the nucleophile Y:⊖ at the carbon bearing the halogen substituent to form an intermediate carbanion 4 (Equation 14-3). The aromatic system is destroyed on forming the anion, and the carbon at the reaction site changes from planar (sp2 bonds) to tetrahedral (sp3 bonds).
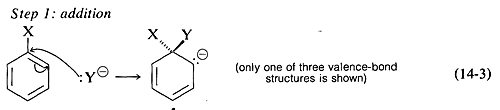
In the second step, loss of an anion, X⊖ or Y⊖, regenerates an aromatic system, and, if X⊖ is lost, the overall reaction is nucleophilic displacement of X by Y (Equation 14-4).
In the case of a neutral nucleophilic reagent, Y or HY, the reaction sequence would be the same except for the necessary adjustments in the charge of the intermediate:
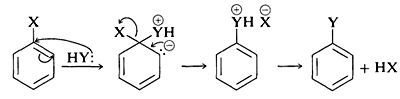
Why is this reaction pathway generally unfavorable for the simple aryl halides? The answer is that the intermediate 4, which we can express as a hybrid of the valence-bond structures 4a-4c, is too high in energy to be formed at any practical rate. Not only has 4 lost the aromatic stabilization of the benzene ring, but its formation results in transfer of negative charge to the ring carbons, which themselves are not very electronegative:

However, when strongly electron-attracting groups are located on the ring at the ortho-para positions, the intermediate anion is stabilized by delocalization of electrons from the ring carbons to more favorable locations on the substituent groups. As an example, consider the displacement of bromine by OCH3 in the reaction of 4-bromonitrobenzene and methoxide ion:
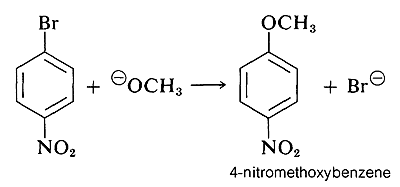
The anionic intermediate formed by addition of methoxide ion to the aryl halide can be described by the valence-bond structures 5a-5d. Of these structures 5d is especially important because in it the charge is transferred from the ring carbons to the oxygen of the nitro substituent:
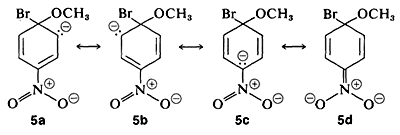
Substituents in the meta positions have much less effect on the reactivity of an aryl halide because delocalization of electrons to the substituent is not possible. No formulas can be written analogous to 5c and 5d in which the negative charges are both on atoms next to positive nitrogen, 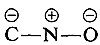 and
and 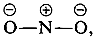
and 
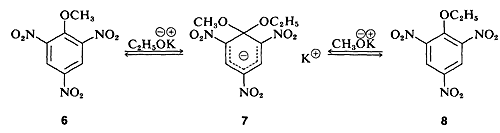
In a few instances, stable compounds resembling the postulated reaction intermediate have been isolated. One classic example is the complex 7 (isolated by J. Meisenheimer), which is the product of the reaction of either the methyl aryl ether 6 with potassium ethoxide, or the ethyl aryl ether 8 and potassium methoxide:
Exercises
Questions
Q16.7.1
Propose a mechanism for the following reaction:

Solutions
S16.7.1

Sir , will a HF molecule be formed at the end?
ReplyDelete