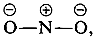Haloform reaction
NETT REACTION :
Haloform Reaction is a type of organic reaction where haloform is produced by halogenation of methyl ketone in the presence of a base. It is a kind of nuclophilic substitution reaction. This reaction is used in quanitatve analysis to indicate the presence of methyl ketone.


NETT REACTION :
Haloform Reaction is a type of organic reaction where haloform is produced by halogenation of methyl ketone in the presence of a base. It is a kind of nuclophilic substitution reaction. This reaction is used in quanitatve analysis to indicate the presence of methyl ketone.
If the hypohalite used in this reaction is sodium hypoiodite, the product is iodoform (CHI3). Such a haloform reaction is also known as iodoform test. As iodoform is a yellow crystalline solid with a characteristic odor and melting point it can be handled much more easily handled in the the laboratory.
- When methyl ketones are treated with the halogen in basic solution, polyhalogenaton followed by cleavage of the methyl group occurs.
- The products are the carboxylate and trihalomethane, otherwise known as haloform.
- The reaction proceeds via successively faster halogenations at the α-position until the 3 H have been replaced.
- The halogenations get faster since the halogen stablises the enolate negative charge and makes it easier to form.
- Then a nucleophilic acyl substitution by hydroxide displaces the anion CX3 as a leaving group that rapidly protonates.
- This reaction is often performed using iodine and as a chemical test for identifying methyl ketones. Iodoform is yellow and precipitates under the reaction conditions.
| MECHANISM OF THE HALOFORM REACTION OF METHYL KETONES | |
Step 1: First, an acid-base reaction. Hydroxide functions as a base and removes the acidic α-hydrogen giving the enolate. | |
| Step 2: The nucleophilic enolate reacts with the iodine giving the halogenated ketone and an iodide ion. | |
Step 3: Steps 1 and 2 repeat twice more yielding the trihalogenated ketone. | |
| Step 4: The hydroxide now reacts as a nucleophile at the electrophilic carbonyl carbon, with the C=O becoming a C-O single bond and the oxygen is now anionic.  | |
| Step 5: Reform the favourable C=O and displace a leaving group, the trihalomethyl system which is stabilised by the 3 halogens. This gives the carboxylic acid. | |
| Step 6: An acid-base reaction. The trihalomethyl anion is protonated by the carboxylic acid, giving the carboxylate and the haloform (trihalomethane).  | |































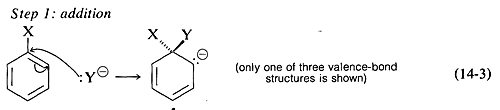
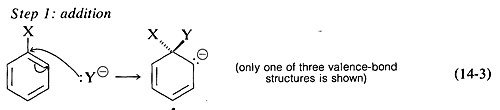
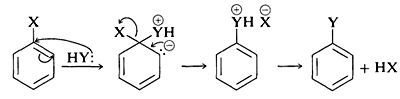

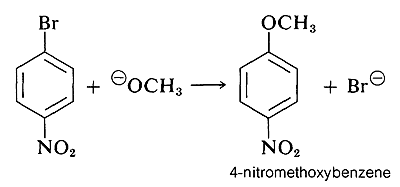
 and
and