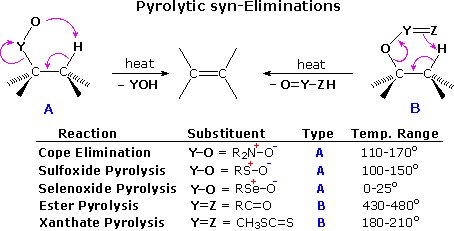
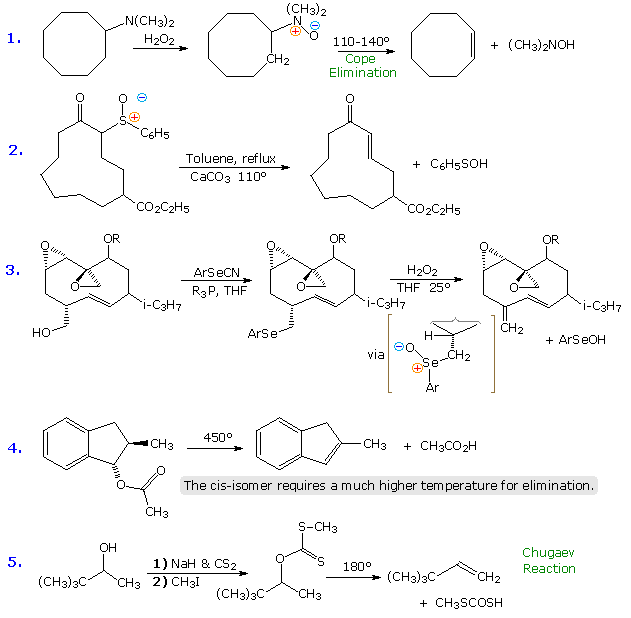
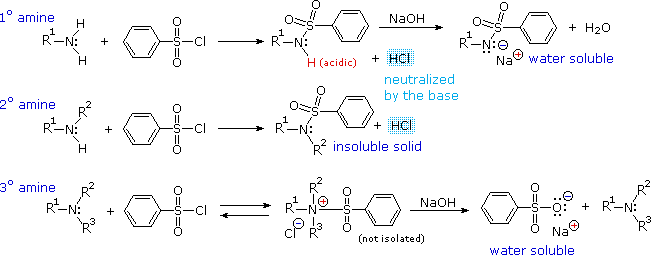
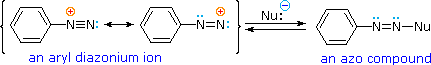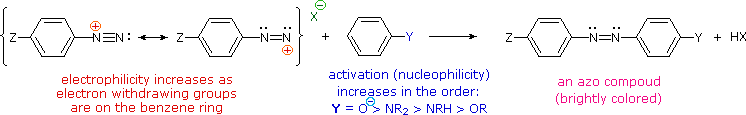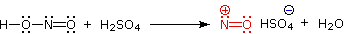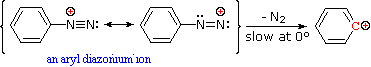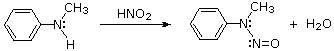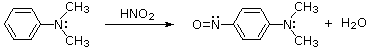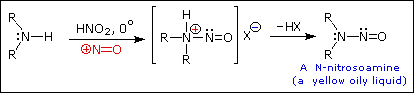Substitution with Loss of Nitrogen
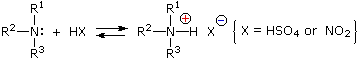
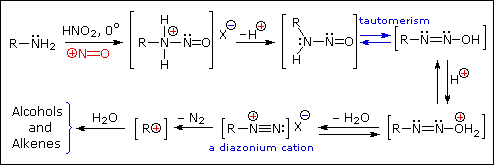
Aryl diazonium salts are important intermediates. They are prepared in cold (0 º to 10 ºC) aqueous solution, and generally react with nucleophiles with loss of nitrogen. Some of the more commonly used substitution reactions are shown in the following diagram. Since the leaving group (N2) is thermodynamically very stable, these reactions are energetically favored. Those substitution reactions that are catalyzed by cuprous salts are known as Sandmeyer reactions. Fluoride substitution occurs on treatment with BF4(–), a reaction known as theSchiemann reaction. Stable diazonium tetrafluoroborate salts may be isolated, and on heating these lose nitrogen to give an arylfluoride product. The top reaction with hypophosphorus acid, H3PO2, is noteworthy because it achieves the reductive removal of an amino (or nitro) group. Unlike the nucleophilic substitution reactions, this reduction probably proceeds by a radical mechanism.'
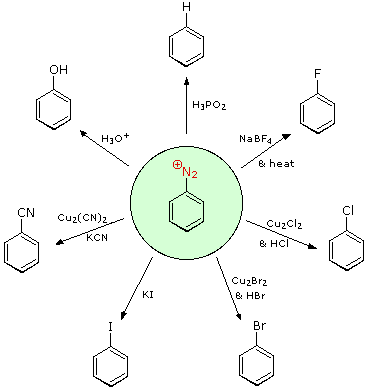
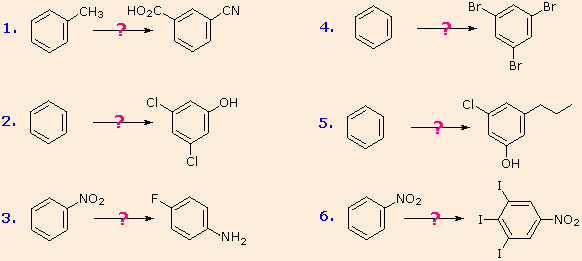
These aryl diazonium substitution reactions significantly expand the tactics available for the synthesis of polysubstituted benzene derivatives. Consider the following options:
(i) The usual precursor to an aryl amine is the corresponding nitro compound. A nitro substituent deactivates an aromatic ring and directs electrophilic substitution to meta locations.
(ii) Reduction of a nitro group to an amine may be achieved in several ways. The resulting amine substituent strongly activates an aromatic ring and directs electrophilic substitution to ortho & para locations.
(iii) The activating character of an amine substituent may be attenuated by formation of an amide derivative (reversible), or even changed to deactivating and meta-directing by formation of a quaternary-ammonium salt (irreversible).
(iv) Conversion of an aryl amine to a diazonium ion intermediate allows it to be replaced by a variety of different groups (including hydrogen), which may in turn be used in subsequent reactions.
The following examples illustrate some combined applications of these options to specific cases. You should try to conceive a plausible reaction sequence for each. Once you have done so, you may check suggested answers by clicking on the question mark for each.
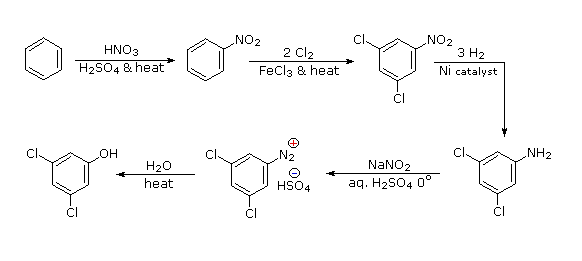
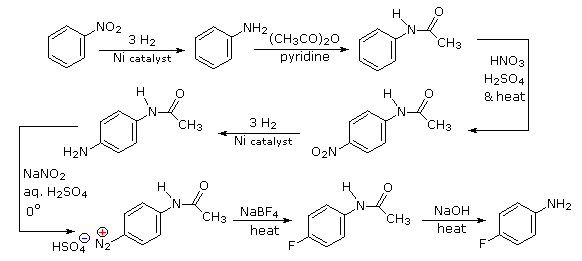
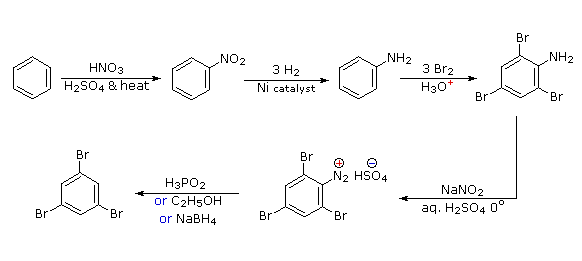
Polybromination of benzene would lead to ortho/para substitution. In order to achieve the mutual meta-relationship of three bromines, it is necessary to introduce a powerful ortho/para-directing prior to bromination, and then remove it following the tribromination. An amino group is ideal for this purpose. Reductive removal of the diazonium group may be accomplished in several ways (three are shown).
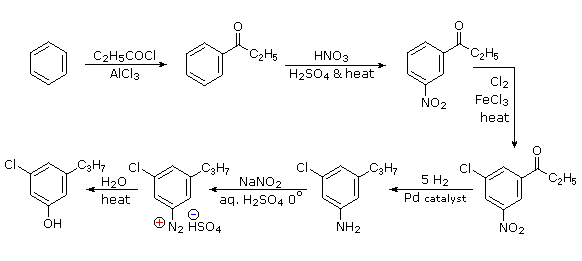
The propyl substituent is best introduced by Friedel-Crafts acylation followed by reduction, and this cannot be carried out in the presence of a nitro substituent. Since an acyl substituent is a meta-director, it is logical to use this property to locate the nitro and chloro groups before reducing the carbonyl moiety. The same reduction method can be used to reduce both the nitro group (to an amine) and the carbonyl group to propyl. We have already seen the use of diazonium intermediates as precursors to phenols.
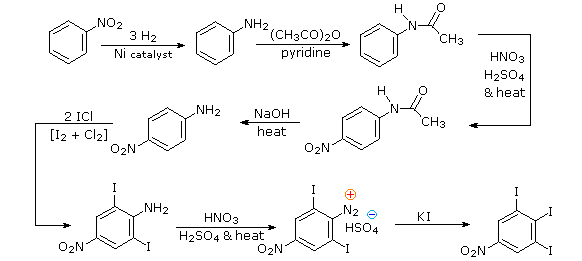
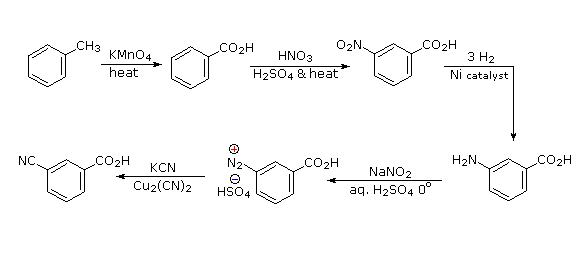
Aromatic iodination can only be accomplished directly on highly activated benzene compounds, such as aniline, or indirectly by way of a diazonium intermediate. Once again, a deactivated amino group is the precursor of p-nitroaniline (prb.#3). This aniline derivative requires the more electrophilic iodine chloride (ICl) for ortho-iodination because of the presence of a deactivating nitro substituent. Finally, the third iodine is introduced by the diazonium ion procedure.
Bonding to Nitrogen
A resonance description of diazonium ions shows that the positive charge is delocalized over the two nitrogen atoms. It is not possible for nucleophiles to bond to the inner nitrogen, but bonding (or coupling) of negative nucleophiles to the terminal nitrogen gives neutral azo compounds. As shown in the following equation, this coupling to the terminal nitrogen should be relatively fast and reversible. The azo products may exist as E / Z stereoisomers. In practice it is found that the E-isomer predominates at equilibrium.
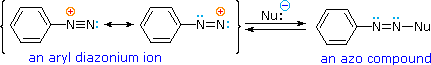
Unless these azo products are trapped or stabilized in some manner, reversal to the diazonium ion and slow nucleophilic substitution at carbon (with irreversible nitrogen loss) will be the ultimate course of reaction, as described in the previous section. For example, if phenyldiazonium bisufate is added rapidly to a cold solution of sodium hydroxide a relatively stable solution of sodium phenyldiazoate (the conjugate base of the initially formed diazoic acid) is obtained. Lowering the pH of this solution regenerates phenyldiazoic acid (pKa ca. 7), which disassociates back to the diazonium ion and eventually undergoes substitution, generating phenol.
| C6H5N2(+) HSO4(–) + NaOH (cold solution) |  | C6H5N2–OH + NaOH (cold) | | C6H5N2–O(–) Na(+) |
| phenyldiazonium bisulfate | | phenyldiazoic acid | | sodium phenyldiazoate |
Aryl diazonium salts may be reduced to the corresponding hydrazines by mild reducing agents such as sodium bisulfite, stannous chloride or zinc dust. The bisulfite reduction may proceed by an initial sulfur-nitrogen coupling, as shown in the following equation.
Ar-N2(+) X(–) | NaHSO3
 |
Ar-N=N-SO3H | NaHSO3
|
Ar-NH-NH-SO3H | H2O
|
Ar-NH-NH2 + H2SO4 |
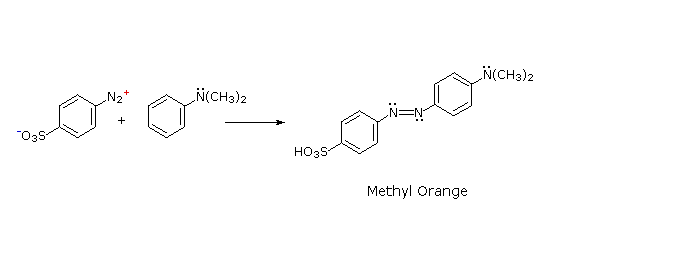
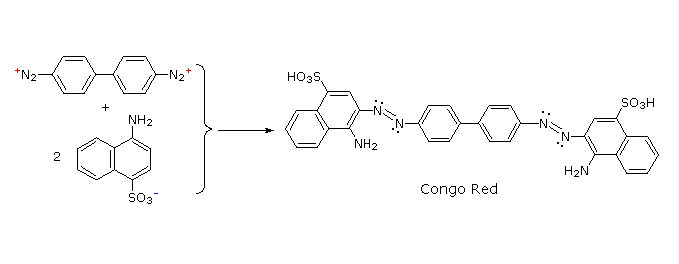
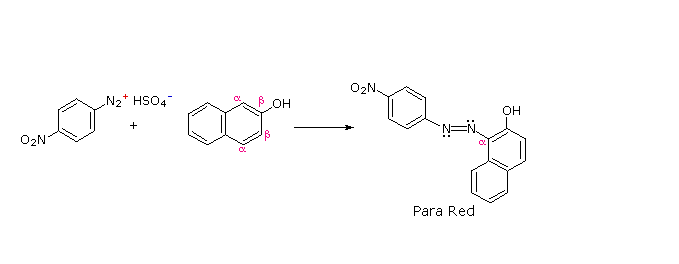
The most important application of diazo coupling reactions is electrophilic aromatic substitution of activated benzene derivatives by diazonium electrophiles. The products of such reactions are highly colored aromatic azo compounds that find use as synthetic dyestuffs, commonly referred to as azo dyes. Azobenzene (Y=Z=H) is light orange; however, the color of other azo compounds may range from red to deep blue depending on the nature of the aromatic rings and the substituents they carry. Azo compounds may exist as cis/trans isomer pairs, but most of the well-characterized and stable compounds are trans.
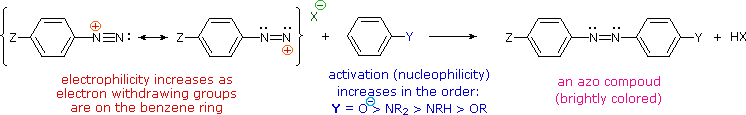
Some examples of azo coupling reactions are shown below. A few simple rules are helpful in predicting the course of such reactions:
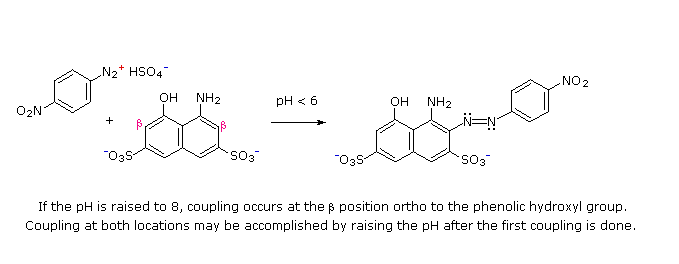
(i) At acid pH (< 6) an amino group is a stronger activating substituent than a hydroxyl group (i.e. a phenol). At alkaline pH (> 7.5) phenolic functions are stronger activators, due to increased phenoxide base concentration.
(ii) Coupling to an activated benzene ring occurs preferentially para to the activating group if that location is free. Otherwise ortho-coupling will occur.
(iii) Naphthalene normally undergoes electrophilic substitution at an alpha-location more rapidly than at beta-sites; however, ortho-coupling is preferred. See the diagram for examples of α / β notation in naphthalenes.
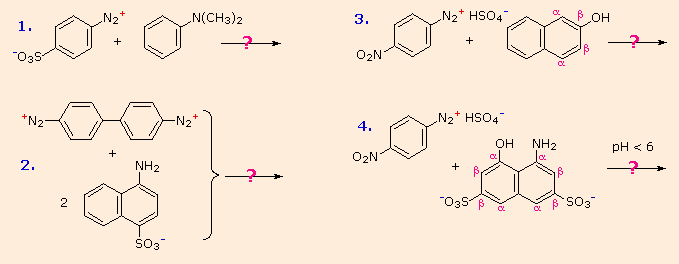
You should try to conceive a plausible product structure for each of the following couplings.