Properties of Amines
amines - nomenclature
Amines are derivatives of ammonia in which one or more of
the hydrogens has been replaced by
an alkyl or aryl group. The nomenclature of amines is complicated by the fact
that several different nomenclature systems exist, and there is no clear
preference for one over the others.
Furthermore, the terms primary (1º),
secondary (2º) &
tertiary (3º) are used to classify amines in a completely
different manner than they were used for
alcohols or alkyl
halides.
When applied to amines these
terms refer to the
number of alkyl (or aryl) substituents bonded to the nitrogen
atom, whereas in other cases they refer to the nature of an
alkyl group.
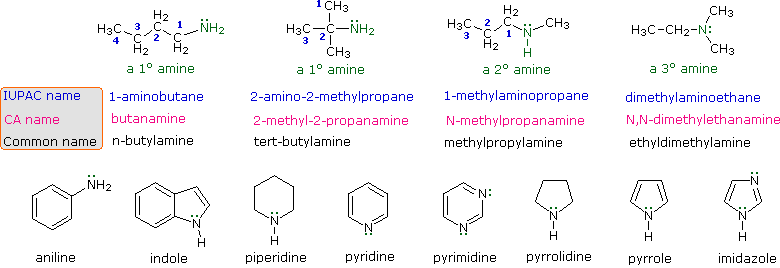
The four compounds shown in the top row of the
following diagram are all C4H11N isomers.
The first two
are classified as 1º-amines, since only one alkyl group is bonded to the
nitrogen; however, the alkyl group is
primary in the first example and
tertiary in the second. The
third and fourth compounds in the row are 2º and 3º-amines
respectively.
A
nitrogen bonded to four alkyl groups
will necessarily be
positively charged, and is called a 4º-ammonium cation.
Forexample, (CH3)4N(+) Br(–) is
tetramethylammonium
bromide.
Amines are derivatives of ammonia in which one or more of
the hydrogens has been replaced by
an alkyl or aryl group. The nomenclature of amines is complicated by the fact
that several different nomenclature systems exist, and there is no clear
preference for one over the others.
Furthermore, the terms primary (1º),
secondary (2º) &
tertiary (3º) are used to classify amines in a completely
different manner than they were used for
alcohols or alkyl
halides.
When applied to amines these
terms refer to the
number of alkyl (or aryl) substituents bonded to the nitrogen
atom, whereas in other cases they refer to the nature of an
alkyl group.
The four compounds shown in the top row of the
following diagram are all C4H11N isomers.
The first two
are classified as 1º-amines, since only one alkyl group is bonded to the
nitrogen; however, the alkyl group is
primary in the first example and
tertiary in the second. The
third and fourth compounds in the row are 2º and 3º-amines
respectively.
A
nitrogen bonded to four alkyl groups
will necessarily be
positively charged, and is called a 4º-ammonium cation.
Forexample, (CH3)4N(+) Br(–) is
tetramethylammonium
bromide.
Structure
The Nitrogen is SP3 hybridised but
rapidly inverting, for NH3 the inversion rate is = 2 x 1011;

The
fact this rate of inversion is so regular is exploited in the ammonia clock. An
important implication of this is that chiral amines racemise rapidly. This can
be stopped by use of a rigid framework, an example of which is
1-aza-bicyclo-[2,2,2]-heptane;
The Nitrogen is SP3 hybridised but
rapidly inverting, for NH3 the inversion rate is = 2 x 1011;
The
fact this rate of inversion is so regular is exploited in the ammonia clock. An
important implication of this is that chiral amines racemise rapidly. This can
be stopped by use of a rigid framework, an example of which is
1-aza-bicyclo-[2,2,2]-heptane;
Boiling Point and Water Solubility
It is instructive to compare the boiling points and water solubility of
amines with those of corresponding alcohols and ethers.
The dominant factor here is hydrogen bonding, and the first table
below documents the powerful intermolecular attraction that results from -O-H---O- hydrogen bonding in alcohols (light blue columns).
Corresponding -N-H---N- hydrogen bonding is weaker, as the
lower boiling points of similarly sized amines (light green columns)
demonstrate. Alkanes provide reference compounds in which hydrogen bonding is
not possible, and the increase in boiling point for equivalent 1º-amines is
roughly half the increase observed for equivalent alcohols.
| Compound | CH3CH3 | CH3OH | CH3NH2 | CH3CH2CH3 | CH3CH2OH | CH3CH2NH2 |
|---|---|---|---|---|---|---|
| Mol.Wt. | 30 | 32 | 31 | 44 | 46 | 45 |
| Boiling Point ºC | -88.6º | 65º | -6.0º | -42º | 78.5º | 16.6º |
The second table illustrates differences associated with isomeric 1º, 2º
& 3º-amines, as well as the influence of chain branching.
Since 1º-amines have two hydrogens available for hydrogen bonding, we
expect them to have higher boiling points than isomeric 2º-amines, which in
turn should boil higher than isomeric 3º-amines (no hydrogen bonding).
Indeed, 3º-amines have boiling points similar to equivalent sized ethers;
and in all but the smallest compounds, corresponding ethers, 3º-amines and
alkanes have similar boiling points.
In the examples shown here, it is further demonstrated that chain
branching reduces boiling points by 10 to 15 ºC.
| Compound | CH3(CH2)2CH3 | CH3(CH2)2OH | CH3(CH2)2NH2 | CH3CH2NHCH3 | (CH3)3CH | (CH3)2CHOH | (CH3)2CHNH2 | (CH3)3N |
|---|---|---|---|---|---|---|---|---|
| Mol.Wt. | 58 | 60 | 59 | 59 | 58 | 60 | 59 | 59 |
| Boiling Point ºC | -0.5º | 97º | 48º | 37º | -12º | 82º | 34º | 3º |
The water solubility of 1º and 2º-amines is similar to that of comparable
alcohols.
As expected, the water solubility of 3º-amines and ethers is also
similar.
These comparisons, however, are valid only for pure compounds in neutral
water.
The basicity of amines (next section) allows them to be dissolved
in dilute mineral acid solutions, and this property facilitates their
separation from neutral compounds such as alcohols and hydrocarbons by
partitioning between the phases of non-miscible solvents.
Basicity of Amines
A review of basic acid-base concepts should be
helpful to the following discussion.
Like ammonia, most amines are Brønsted and Lewis bases, but their
base strength can be changed enormously by substituents.
It is common to compare basicity's quantitatively by using thepKa's of their conjugate
acids rather than their pKb's.
Since pKa + pKb = 14, the higher the pKa the stronger the base, in contrast
to the usual inverse relationship of pKa with acidity.
Most simple alkyl amines have pKa's in the range 9.5 to
11.0, and their water solutions are basic (have a pH of 11 to 12, depending on
concentration).
The first four compounds in the following table, including ammonia, fall
into that category.
The last five compounds (colored cells) are significantly weaker bases as
a consequence of three factors. The first of these is the hybridization of the
nitrogen.
In pyridine the nitrogen is sp2 hybridized, and
in nitriles (last entry) an sp hybrid nitrogen is part of the triple
bond.
In each of these compounds (shaded red) the non-bonding electron pair is
localized on the nitrogen atom, but increasing s-character brings it closer to
the nitrogen nucleus, reducing its tendency to bond to a proton.
Compound
|  |  |  | NH3 |  | 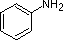 |  | 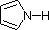 |  | CH3C≡N |
|---|---|---|---|---|---|---|---|---|---|---|
| pKa | 11.0 | 10.7 | 10.7 | 9.3 | 5.2 | 4.6 | 1.0 | 0.0 | -1.0 | -10.0 |
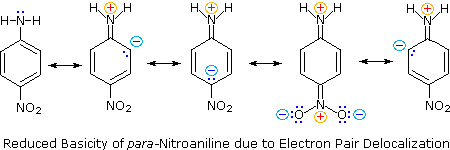
Secondly,
aniline and p-nitroaniline (first two green shaded structures) are weaker bases
due to delocalization of the nitrogen non-bonding electron pair into the aromatic
ring (and the nitro substituent).
This is the
same delocalization that results in activation of a benzene ring toward
electrophilic substitution.
The
following resonance equations, which are similar to those used to explain the enhanced acidity of ortho and
para-nitrophenols illustrate electron pair delocalization in
p-nitroaniline.
Indeed,
aniline is a weaker base than cyclohexyl amine by roughly a million fold, the
same factor by which phenol is a stronger acid than cyclohexanol.
This
electron pair delocalization is accompanied by a degree of rehybridization of
the amino nitrogen atom, but the electron pair delocalization is probably the
major factor in the reduced basicity of these compounds.
A
similar electron pair delocalization is responsible for the very low basicity
(and nucleophilic reactivity) of amide nitrogen atoms (last green shaded
structure).
This
feature was instrumental in moderating the influence of amine substituents on
aromatic ring substitution, and will be discussed further in the section
devoted to carboxylic acid derivatives.
Conjugated amine groups
influence the basicity of an existing amine.
Although
4-dimethylaminopyridine (DMAP) might appear to be a base similar in strength to
pyridine or N,N-dimethylaniline, it is actually more than ten thousand times
stronger, thanks to charge delocalization in its conjugate acid.
The structure in the
gray box shows the locations over which positive charge (colored red) is
delocalized in the conjugate acid. This compound is often used as a catalyst
for acyl transfer reactions.
Finally, the very low basicity of pyrrole (shaded blue) reflects the exceptional delocalization of the nitrogen electron pair associated with its incorporation in an aromatic ring.
Indole (pKa = -2) and imidazole (pKa = 7.0), see above, also have similar
heterocyclic aromatic rings.
Imidazole is over
a million times more basic than pyrrole because the
sp2 nitrogen that is part of one double bond is
structurally similar to pyridine, and has a comparable basicity.
Although resonance
delocalization generally reduces the basicity of amines, a dramatic example of
the reverse effect is found in the compound guanidine (pKa = 13.6).
Here, as shown below,
resonance stabilization of the base is small, due to charge separation, while
the conjugate acid is stabilized strongly by charge delocalization.
Consequently,
aqueous solutions of guanidine are nearly as basic as are solutions of sodium
hydroxide.

The relationship of
amine basicity to the acidity of the corresponding conjugate acids may be
summarized in a fashion analogous to that noted earlier for acids:
Strong
bases have weak conjugate acids, and weak bases have strong
conjugate
acids.
Acidity of Amines
We normally
think of amines as bases, but it must be remembered that 1º and 2º-amines are
also very weak acids (ammonia has a pKa = 34).
In this
respect it should be noted that pKa is being used as a
measure of the acidity of the amine itself rather than its conjugate acid, as
in the previous section.
For ammonia
this is expressed by the following hypothetical equation:
NH3 + H2O à NH2(–) + H2O-H(+)
The same
factors that decreased the basicity of amines increase their acidity.
This is
illustrated by the following examples, which are shown in order of increasing
acidity.
It
should be noted that the first four examples have the same order and degree of
increased acidity as they exhibited decreased basicity in the previous
table.
The first
compound is a typical 2º-amine, and the three next to it are characterized by
varying degrees of nitrogen electron pair delocalization.
The
last two compounds (shaded blue) show the influence of adjacent sulfonyl and
carbonyl groups on N-H acidity.
From
previous discussion it should be clear that the basicity of these nitrogens is
correspondingly reduced.
| Compound | | | | | C6H5SO2NH2 | 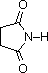 |
|---|---|---|---|---|---|---|
| pKa | 33 | 27 | 19 | 15 | 10 | 9.6 |
The acids
shown here may be converted to their conjugate bases by reaction with bases
derived from weaker acids (stronger bases).
Three
examples of such reactions are shown below, with the acidic hydrogen colored
red in each case.
For
complete conversion to the conjugate base, as shown, a reagent base roughly a
million times stronger is required.
C6H5SO2NH2 +
KOH
|
a sulfonamide base
|
(CH3)3COH +
NaH
|
an alkoxide base
|
(C2H5)2NH + C4H9Li
|
an amide base
|
Important Reagent Bases
The
significance of all these acid-base relationships to practical organic
chemistry lies in the need for organic bases of varying strength, as reagents
tailored to the requirements of specific reactions.
The common
base sodium hydroxide is not soluble in many organic solvents, and is therefore
not widely used as a reagent in organic reactions.
Most base
reagents are alkoxide salts, amines or amide salts. Since alcohols are much
stronger acids than amines, their conjugate bases are weaker than amide bases,
and fill the gap in base strength between amines and amide salts.
In
the following table, pKa again refers to the conjugate
acid of the base drawn above it.
| Base Name | Pyridine | Triethyl Amine | Hünig's Base | Barton's Base | Potassium t-Butoxide | Sodium HMDS | LDA |
|---|---|---|---|---|---|---|---|
| Formula | | (C2H5)3N | 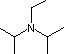 | 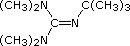 | (CH3)3CO(–) K(+) | [(CH3)3Si]2N(–) Na(+) | [(CH3)2CH]2N(–) Li(+) |
| pKa | 5.3 | 10.7 | 11.4 | 14 | 19 | 26 | 35.7 |
Pyridine is
commonly used as an acid scavenger in reactions that produce mineral acid
co-products.
Its
basicity and nucleophilicity may be modified by steric hindrance, as in the
case of 2,6-dimethylpyridine (pKa=6.7), or resonance
stabilization, as in the case of 4-dimethylaminopyridine (pKa=9.7).
Hünig's
base is relatively non-nucleophilic (due to steric hindrance), and like DBU is
often used as the base in E2 elimination reactions conducted in non-polar
solvents.
Barton's
base is a strong, poorly-nucleophilic, neutral base that serves in cases where
electrophilic substitution of DBU or other amine bases is a problem.
The
alkoxides are stronger bases that are often used in the corresponding alcohol
as solvent, or for greater reactivity in DMSO.
Finally,
the two amide bases see widespread use in generating enolate bases from
carbonyl compounds and other weak carbon acids.
Preparation of Amines
Preparation of
Primary Amines
Although
direct alkylation of ammonia by alkyl halides leads to 1º-amines, alternative
procedures are preferred in many cases.
These methods require two steps, but they provide pure product, usually in good yield.
Gabriel Synthesis
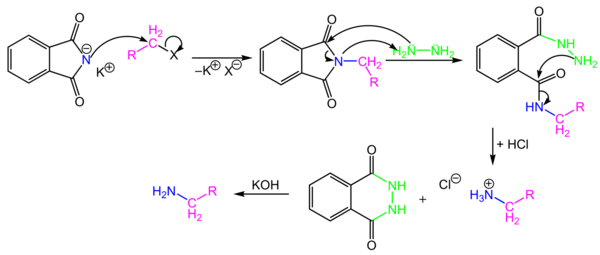
Potassium phthalimide
is a -NH2-synthon which allows the preparation of
primary amines by reaction with alkyl halides. After alkylation, the phthalimid
is not nucleophile and does not react anymore. Product is cleaved by reaction
with base or hydrazine, which leads to a stable cyclic product.
Mechanism of the Gabriel Synthesis
Note: Phthalimide is
acidic!


Cleavage:

The general
strategy is to first form a carbon-nitrogen bond by reacting a nitrogen nucleophile
with a carbon electrophile.
The following table lists several general examples of this strategy in the rough order of decreasing nucleophilicity of the nitrogen reagent.
In the second step, extraneous nitrogen substituents that may have facilitated this bonding are removed to give the amine product.
Nitrogen
Reactant |
Carbon
Reactant |
1st Reaction
Type |
Initial Product
|
2nd Reaction
Conditions |
2nd Reaction
Type |
Final Product
|
|---|---|---|---|---|---|---|
| N3(–) | RCH2-X or R2CH-X | SN2 | RCH2-N3 or R2CH-N3 | LiAlH4 or 4 H2 & Pd | Hydrogenolysis | RCH2-NH2 or R2CH-NH2 |
| C6H5SO2NH(–) | RCH2-X or R2CH-X | SN2 | RCH2-NHSO2C6H5 or R2CH-NHSO2C6H5 | Na in NH3 (liq) | Hydrogenolysis | RCH2-NH2 or R2CH-NH2 |
| CN(–) | RCH2-X or R2CH-X | SN2 | RCH2-CN or R2CH-CN | LiAlH4 | Reduction | RCH2-CH2NH2 or R2CH-CH2NH2 |
| NH3 | RCH=O or R2C=O | Addition / Elimination | RCH=NH or R2C=NH | H2 & Ni or NaBH3CN | Reduction | RCH2-NH2 or R2CH-NH2 |
| NH3 | RCOX | Addition / Elimination | RCO-NH2 | LiAlH4 | Reduction | RCH2-NH2 |
| NH2CONH2 (urea) | R3C(+) | SN1 | R3C-NHCONH2 | NaOH soln. | Hydrolysis | R3C-NH2 |
A specific
example of each general class is provided in the diagram below.
In the first two, an anionic nitrogen species undergoes an SN2 reaction with a modestly electrophilic alkyl halide reactant.
For example #2 an acidic phthalimide derivative of ammonia has been substituted for the sulfonamide analog listed in the table. The principle is the same for the two cases, as will be noted later.
Example #3 is similar in nature, but extends the carbon system by a methylene group (CH2).
In all three of these methods 3º-alkyl halides cannot be used because the major reaction path is an E2 elimination.
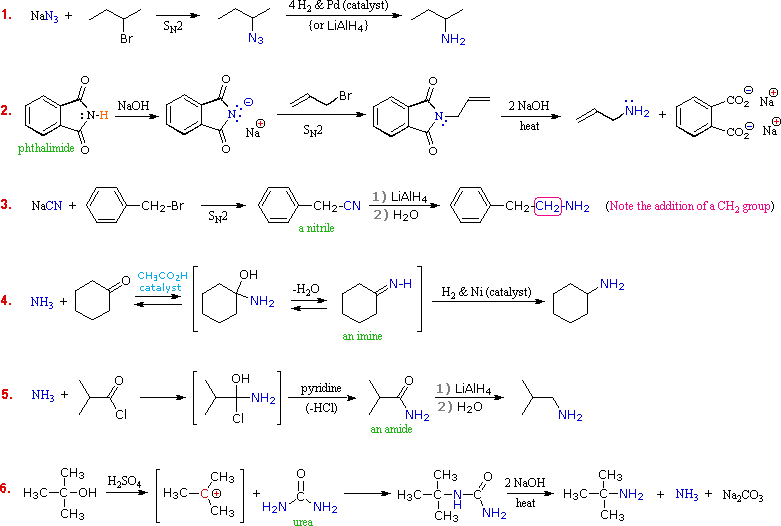
The
methods illustrated by examples #4 and #5 proceed by attack of ammonia, or
equivalent nitrogen nucleophiles, at the electrophilic carbon of a carbonyl
group.
A full
discussion of carbonyl chemistry is presented later, but for present purposes
it is sufficient to recognize that the C=O double bond is polarized so that the
carbon atom is electrophilic.
Nucleophile addition to aldehydes and ketones is often catalyzed by acids. Acid halides and anhydrides are even more electrophilic, and do not normally require catalysts to react with nucleophiles.
The reaction of ammonia with aldehydes or ketones occurs by a reversible addition-elimination pathway to give imines (compounds having a C=N function).
These intermediates are not usually isolated, but are reduced as they are formed (i.e. in situ).
Acid chlorides react with ammonia to give amides, also by an addition-elimination path, and these are reduced to amines by LiAlH4.
The 6th example is a specialized procedure for bonding an amino group to a 3º-alkyl group (none of the previous methods accomplishes this).
Since a carbocation is the electrophilic species, rather poorly nucleophilic nitrogen reactants can be used. Urea, the diamide of carbonic acid, fits this requirement nicely. The resulting 3º-alkyl-substituted urea is then hydrolyzed to give the amine.
One important method of preparing 1º-amines, especially aryl amines, uses a reverse strategy.
Here a strongly electrophilic nitrogen species (NO2(+)) bonds to a nucleophilic carbon compound.
This nitration reaction gives a nitro group that can be reduced to a 1º-amine by any of several reduction procedures.
The
Hofmann rearrangement of 1º-amides provides an additional synthesis of
1º-amines.
Preparation
of Secondary and Tertiary Amines
Of the six
methods described above, three are suitable for the preparation of 2º and/or
3º-amines. These are
1.
Alkylation of the sulfonamide
derivative of a 1º-amine. Gives 2º-amines.
2.
Reduction of alkyl imines and dialkyl
iminium salts. Gives 2º & 3º-amines.
3.
Reduction of amide derivatives of 1º
& 2º-amines. Gives 2º & 3º-amines.
Examples
showing the application of these methods to the preparation of specific amines
are shown in the following diagram.
The sulfonamide procedure used in the first example is similar in concept to the phthalimide example #2 presented in the previous diagram.
In both cases the acidity of the nitrogen reactant (ammonia or amine) is greatly enhanced by conversion to an imide or sulfonamide derivative.
The nucleophilic conjugate base of this acidic nitrogen species is then prepared by treatment with sodium or potassium hydroxide, and this undergoes an SN2 reaction with a 1º or 2º-alkyl halide.
Finally, the activating group is removed by hydrolysis (phthalimide) or reductive cleavage (sulfonamide) to give the desired amine.
The phthalimide method is only useful for preparing 1º-amines, whereas the sulfonamide procedure may be used to make either 1º or 2º-amines.

Examples #2
& #3 make use of the carbonyl reductive amination reaction (method #4 in
the preceding table.
This versatile procedure may be used to prepare all classes of amines (1º, 2º & 3º), as shown here and above.
A weak acid catalyst is necessary for imine formation, which takes place by amine addition to the carbonyl group, giving a 1-aminoalcohol intermediate, followed by loss of water.
The final reduction of the C=N double bond may be carried out catalytically (Pt & Pd catalysts may be used instead of Ni) or chemically (by NaBH3CN).
The imine or enamine intermediates are normally not isolated, but are immediately reduced to the amine product.
Another
general method for preparing all classes of amines makes use of amide
intermediates, easily made from ammonia or amines by reaction with carboxylic
acid chlorides or anhydrides.
.
Examples #4 & #5 illustrate applications of this method. As with the previous method, 1º-amines give 2º-amine products, and 2º-amines give 3º-amine products. The last example (#6) shows how 4º-ammonium salts may be prepared by repeated (exhaustive) alkylation of amines.
Leuckart reaction

reaction of amines
Electrophilic
Substitution at Nitrogen
Ammonia
and many amines are not only bases in the Brønsted sense, they are also
nucleophiles that bond to and form products with a variety of electrophiles.
A general equation for such electrophilic substitution of nitrogen is:
2 R2ÑH
+ E(+) à R2NHE(+)  R2ÑE + H(+) (bonded to a base)
R2ÑE + H(+) (bonded to a base)
A list of some electrophiles that are known to react with amines is shown here.
In each case the electrophilic atom or site is colored red.
Electrophile
RCH2–X
RCH2–OSO2R
R2C=O
R(C=O)X
RSO2–Cl
HO–N=O
Name
Alkyl Halide
Alkyl Sulfonate
Aldehyde
or Ketone
Acid Halide
or Anhydride
Sulfonyl Chloride
Nitrous Acid
Ammonia
and many amines are not only bases in the Brønsted sense, they are also
nucleophiles that bond to and form products with a variety of electrophiles.
A general equation for such electrophilic substitution of nitrogen is:
2 R2ÑH
+ E(+) à R2NHE(+)
|
A list of some electrophiles that are known to react with amines is shown here. In each case the electrophilic atom or site is colored red.
Electrophile
|
RCH2–X
|
RCH2–OSO2R
|
R2C=O
|
R(C=O)X
|
RSO2–Cl
|
HO–N=O
|
Name
|
Alkyl Halide
|
Alkyl Sulfonate
|
Aldehyde
or Ketone |
Acid Halide
or Anhydride |
Sulfonyl Chloride
|
Nitrous Acid
|
Alkylation
The hydrogen bromide produced in the reaction combines with some of the
excess ammonia, giving ammonium bromide as a by-product.
Water does not normally react with 1º-alkyl halides to give alcohols, so the
enhanced nucleophilicity of nitrogen relative to oxygen is clearly demonstrated.
2 RCH2Br + NH3 (large
excess)  RCH2NH2 + NH4(+) Br(–)
RCH2NH2 + NH4(+) Br(–)
I amines react with alkyl halides directly to give N-alkylated products. Since
this reaction produces HBr as a co-product, hydrobromide salts of the alkylated
amine or unreacted starting amine (in equilibrium) will also be formed.
2 RNH2 + C2H5Br
à RNHC2H5 +
RNH3(+) Br(–) à RNH2C2H5(+) Br(–) +
RNH2
Unfortunately, the direct alkylation of 1º or 2º-amines to give a more
substituted product does not proceed cleanly.
If a 1:1 ratio of amine to alkyl halide is used, only 50% of the amine will
react because the remaining amine will be tied up as an ammonium halide salt
(remember that one equivalent of the strong acid HX is produced).
If a 2:1 ratio of amine to alkylating agent is used, as in the above equation,
the HX issue is solved, but another problem arises.
Both the starting amine and the product amine are nucleophiles.
Consequently, once the reaction has started, the product amine competes with
the starting material in the later stages of alkylation, and some higher
alkylated products are also formed.
Even 3º-amines may be alkylated to form quaternary (4º) ammonium salts.
When tetraalkyl ammonium salts are desired, as shown in the following example,
Hünig's base may be used to scavenge the HI produced in the three SN2
reactions.
Steric hindrance prevents this 3º-amine (Hünig's base) from being methylated.
C6H5NH2 +
3 CH3I + Hünig's base à C6H5N(CH3)3(+) I(–) +
HI salt of Hünig's base
The hydrogen bromide produced in the reaction combines with some of the excess ammonia, giving ammonium bromide as a by-product.
Water does not normally react with 1º-alkyl halides to give alcohols, so the enhanced nucleophilicity of nitrogen relative to oxygen is clearly demonstrated.
2 RCH2Br + NH3 (large
excess)
|
I amines react with alkyl halides directly to give N-alkylated products. Since this reaction produces HBr as a co-product, hydrobromide salts of the alkylated amine or unreacted starting amine (in equilibrium) will also be formed.
2 RNH2 + C2H5Br
à RNHC2H5 +
RNH3(+) Br(–) à RNH2C2H5(+) Br(–) +
RNH2
|
Unfortunately, the direct alkylation of 1º or 2º-amines to give a more substituted product does not proceed cleanly.
If a 1:1 ratio of amine to alkyl halide is used, only 50% of the amine will react because the remaining amine will be tied up as an ammonium halide salt (remember that one equivalent of the strong acid HX is produced).
If a 2:1 ratio of amine to alkylating agent is used, as in the above equation, the HX issue is solved, but another problem arises.
Both the starting amine and the product amine are nucleophiles. Consequently, once the reaction has started, the product amine competes with the starting material in the later stages of alkylation, and some higher alkylated products are also formed.
Even 3º-amines may be alkylated to form quaternary (4º) ammonium salts.
When tetraalkyl ammonium salts are desired, as shown in the following example, Hünig's base may be used to scavenge the HI produced in the three SN2 reactions.
Steric hindrance prevents this 3º-amine (Hünig's base) from being methylated.
C6H5NH2 +
3 CH3I + Hünig's base à C6H5N(CH3)3(+) I(–) +
HI salt of Hünig's base
|
The Hinsberg Test: Reaction with
benzenesulfonyl chloride
Another electrophilic reagent,
benzenesulfonyl chloride, reacts with amines in a fashion that provides a
useful test for distinguishing primary, secondary and tertiary amines (the
Hinsberg test).
As shown in the following equations, 1º and 2º-amines react to give sulfonamide
derivatives with loss of HCl, whereas 3º-amines do not give any isolable
products other than the starting amine.
In the latter case a quaternary "onium" salt may be formed as an
intermediate, but this rapidly breaks down in water to liberate the original
3º-amine (lower right equation).
As shown in the following equations, 1º and 2º-amines react to give sulfonamide derivatives with loss of HCl, whereas 3º-amines do not give any isolable products other than the starting amine.
In the latter case a quaternary "onium" salt may be formed as an intermediate, but this rapidly breaks down in water to liberate the original 3º-amine (lower right equation).
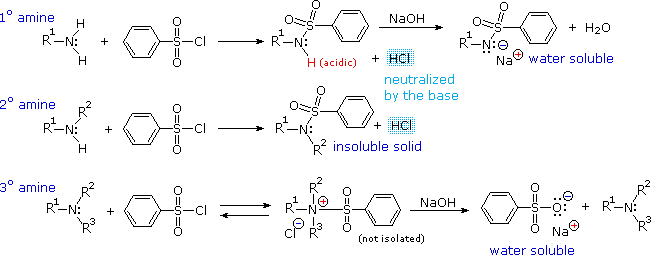
The Hinsberg test is conducted in aqueous base (NaOH or KOH), and the benzenesulfonyl chloride reagent is present as an insoluble oil.
Because of the heterogeneous nature of this system, the rate at which the sulfonyl chloride reagent is hydrolyzed to its sulfonate salt in the absence of amines is relatively slow.
The amine dissolves in the reagent phase, and immediately reacts (if it is 1º or 2º), with the resulting HCl being neutralized by the base.
The sulfonamide derivative from 2º-amines is usually an insoluble solid.
However, the sulfonamide derivative from 1º-amines is acidic and dissolves in the aqueous base. Acidification of this solution then precipitates the sulfonamide of the 1º-amine.
Reaction of Amines with Nitrous Acid
1. 1. Primary Amines
2. 2. Secondary Amines
3. 3. Aryl Amines
1. 3.1. 2º-Aryl Amines:
2. 3.2. 3º-Aryl Amines:
4.
Nitrous acid (HNO2 or HONO) reacts with aliphatic amines in a fashion
that provides a useful test for distinguishing primary, secondary and tertiary
amines.
Nitrous acid is a Brønsted acid of moderate strength (pKa = 3.3).
Because it is unstable, it is prepared immediately before use in the following manner:

Under the acidic conditions of this reaction, all amines undergo reversible salt formation:
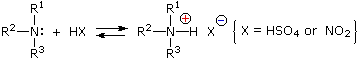
This happens with 3º-amines, and the salts are usually soluble in water.
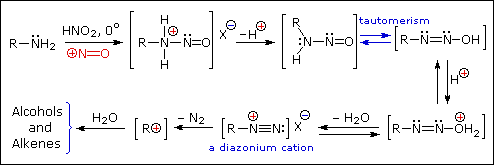
The reactions of nitrous acid with 1°- and 2°- aliphatic amines may be
explained by considering their behavior with the nitrosonium
cation, NO(+), an electrophilic species present in acidic nitrous acid solutions.
Primary Amines
Secondary Amines
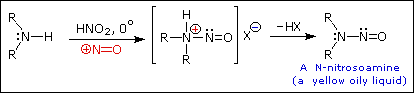
The SN1 product mixtures from 1º-amines are difficult to control, and
rearrangement is common when branched primary alkyl groups are
involved.
The N-nitrosamines formed from 2º-amines are carcinogenic
Aryl Amines
Nitrous acid reactions of 1º-aryl amines generate relatively stable diazonium species that serve as intermediates for a variety of aromatic substitution reactions.
Diazonium cations may be described by resonance contributors, as in the bracketed formulas shown below.
The left-hand contributor is dominant because it has greater bonding.
Loss of nitrogen is slower than in aliphatic 1º-amines because the C-N bond is stronger, and aryl carbocations are comparatively unstable.
Diazonium cations may be described by resonance contributors, as in the bracketed formulas shown below.
The left-hand contributor is dominant because it has greater bonding.
Loss of nitrogen is slower than in aliphatic 1º-amines because the C-N bond is stronger, and aryl carbocations are comparatively unstable.
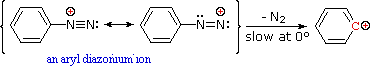
Aqueous solutions of these diazonium ions have sufficient stability at 0º to 10 ºC that they may be used as intermediates in a variety of nucleophilic substitution reactions.
For example, if water is the only nucleophile available for reaction, phenols are formed in good yield.
2º-Aryl Amines:
2º-Aryl amines give N-nitrosamine derivatives on reaction with nitrous acid, and thus behave identically to their aliphatic counterparts.
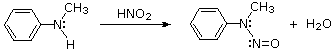
3º-Aryl Amines:
Depending on ring substitution, 3º-Aryl amines may undergo aromatic ring nitrosation at sites ortho or para to the amine substituent.
The nitrosonium cation is not sufficiently electrophilic to react with benzene itself, or even toluene, but highly activated aromatic rings such as amines and phenols are capable of substitution.
Of course, the rate of reaction of NO(+) directly at nitrogen is greater than that of ring substitution, as shown in the previous example.
Once nitrosated, the activating character of the amine nitrogen is greatly diminished; and N-nitrosoaniline derivatives, or indeed any amide derivatives, do not undergo ring nitrosation.
The nitrosonium cation is not sufficiently electrophilic to react with benzene itself, or even toluene, but highly activated aromatic rings such as amines and phenols are capable of substitution.
Of course, the rate of reaction of NO(+) directly at nitrogen is greater than that of ring substitution, as shown in the previous example.
Once nitrosated, the activating character of the amine nitrogen is greatly diminished; and N-nitrosoaniline derivatives, or indeed any amide derivatives, do not undergo ring nitrosation.
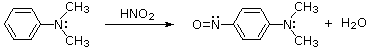
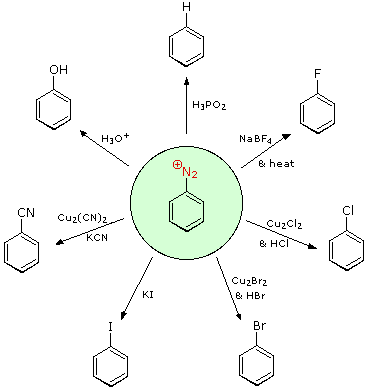
Reactions of Aryl Diazonium Salts
Substitution with Loss of Nitrogen
Aryl diazonium salts are important intermediates.
They are prepared in cold (0 º to 10 ºC) aqueous solution, and generally react with nucleophiles with loss of nitrogen.
Some of the more commonly used substitution reactions are shown in the following diagram.
Since the leaving group (N2) is thermodynamically very stable, these reactions are energetically favored.
Those substitution reactions that are catalyzed by cuprous salts are
known as Sandmeyer reactions.
Fluoride substitution occurs on treatment with BF4(–), a reaction known as theSchiemann reaction.
Stable diazonium tetrafluoroborate salts may be isolated, and on heating these lose nitrogen to give an arylfluoride product.
The top reaction with hypophosphorus acid, H3PO2, is noteworthy because it achieves the reductive removal of an amino (or nitro) group. Unlike the nucleophilic substitution reactions, this reduction probably proceeds by a radical mechanism.'
They are prepared in cold (0 º to 10 ºC) aqueous solution, and generally react with nucleophiles with loss of nitrogen.
Some of the more commonly used substitution reactions are shown in the following diagram.
Since the leaving group (N2) is thermodynamically very stable, these reactions are energetically favored.
Those substitution reactions that are catalyzed by cuprous salts are
known as Sandmeyer reactions.
Fluoride substitution occurs on treatment with BF4(–), a reaction known as theSchiemann reaction.
Stable diazonium tetrafluoroborate salts may be isolated, and on heating these lose nitrogen to give an arylfluoride product.
The top reaction with hypophosphorus acid, H3PO2, is noteworthy because it achieves the reductive removal of an amino (or nitro) group. Unlike the nucleophilic substitution reactions, this reduction probably proceeds by a radical mechanism.'
Consider the following options:
(i) The usual precursor to an
aryl amine is the corresponding nitro compound.
A nitro substituent deactivates
an aromatic ring and directs electrophilic substitution to meta locations.
(ii) Reduction
of a nitro group to an amine may be achieved in several ways. The resulting
amine substituent strongly activates an aromatic ring and directs electrophilic
substitution to ortho & para locations.
(iii) The
activating character of an amine substituent may be attenuated by formation of
an amide derivative (reversible), or even changed to deactivating and
meta-directing by formation of a quaternary-ammonium salt (irreversible).
(iv)
Conversion of an aryl amine to a diazonium ion intermediate allows it to be
replaced by a variety of different groups (including hydrogen), which may in
turn be used in subsequent reactions.
The following examples illustrate some
combined applications of these options to specific cases. You should try to conceive
a plausible reaction sequence for each. Once you have done so, you may check
suggested answers by clicking on the question mark for each.
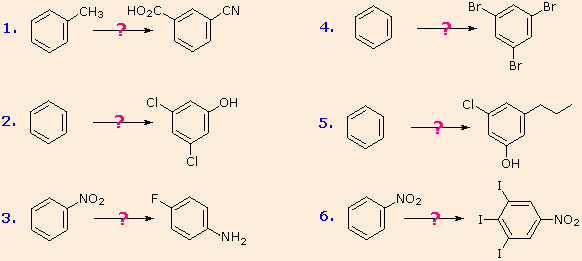
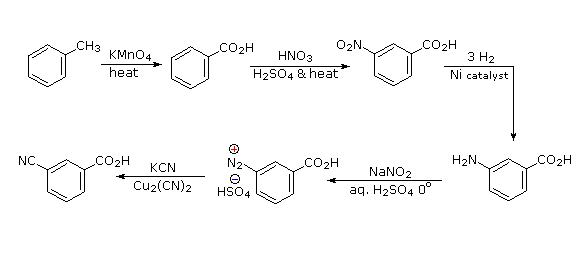
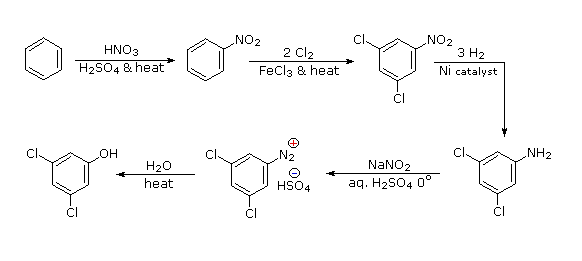
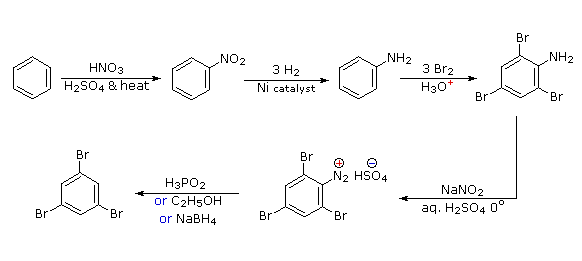
Polybromination of benzene would lead to ortho/para substitution. In order to achieve the mutual meta-relationship of three bromines, it is necessary to introduce a powerful ortho/para-directing prior to bromination, and then remove it following the tribromination.
An amino group is ideal for this purpose. Reductive removal of the diazonium group may be accomplished in several ways (three are shown).
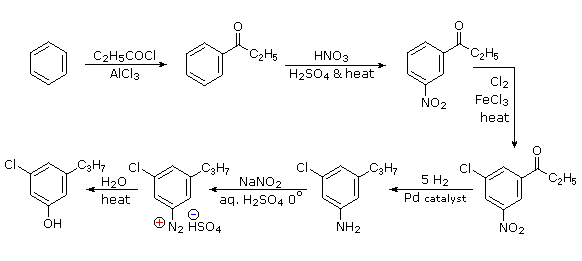
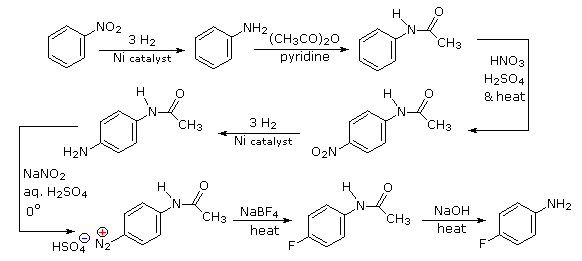
The propyl substituent is best introduced by Friedel-Crafts acylation followed by reduction, and this cannot be carried out in the presence of a nitro substituent. Since an acyl substituent is a meta-director, it is logical to use this property to locate the nitro and chloro groups before reducing the carbonyl moiety. The same reduction method can be used to reduce both the nitro group (to an amine) and the carbonyl group to propyl. We have already seen the use of diazonium intermediates as precursors to phenols.
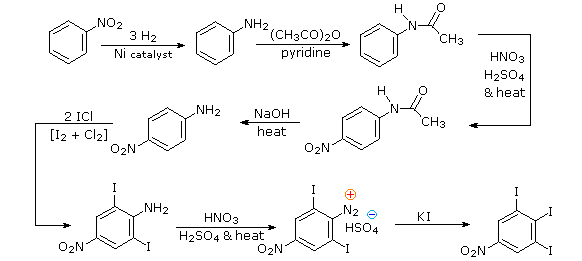
Aromatic iodination can only be accomplished directly on highly activated benzene compounds, such as aniline, or indirectly by way of a diazonium intermediate. Once again, a deactivated amino group is the precursor of p-nitroaniline (prb.#3). This aniline derivative requires the more electrophilic iodine chloride (ICl) for ortho-iodination because of the presence of a deactivating nitro substituent. Finally, the third iodine is introduced by the diazonium ion procedure.
Bonding to Nitrogen
A resonance description of diazonium ions shows that the positive charge is delocalized over the two nitrogen atoms.
It is not possible for nucleophiles to bond to the inner nitrogen, but bonding (or coupling) of negative nucleophiles to the terminal nitrogen gives neutral azo compounds.
As shown in the following equation, this coupling to the terminal nitrogen should be relatively fast and reversible.
The azo products may exist as E / Z stereoisomers. In practice it is found that the E-isomer predominates at equilibrium.
It is not possible for nucleophiles to bond to the inner nitrogen, but bonding (or coupling) of negative nucleophiles to the terminal nitrogen gives neutral azo compounds.
As shown in the following equation, this coupling to the terminal nitrogen should be relatively fast and reversible.
The azo products may exist as E / Z stereoisomers. In practice it is found that the E-isomer predominates at equilibrium.
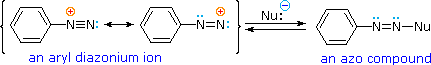
If phenyldiazonium bisufate is added rapidly to a cold solution of sodium hydroxide a relatively stable solution of sodium phenyldiazoate (the conjugate base of the initially formed diazoic acid) is obtained.
Lowering the pH of this solution regenerates phenyldiazoic acid (pKa ca. 7), which disassociates back to the diazonium ion and eventually undergoes substitution, generating phenol.
Lowering the pH of this solution regenerates phenyldiazoic acid (pKa ca. 7), which disassociates back to the diazonium ion and eventually undergoes substitution, generating phenol.
| C6H5N2(+) HSO4(–) + NaOH (cold solution) |  | C6H5N2–OH + NaOH (cold) | | C6H5N2–O(–) Na(+) |
| phenyldiazonium bisulfate | phenyldiazoic acid | sodium phenyldiazoate |
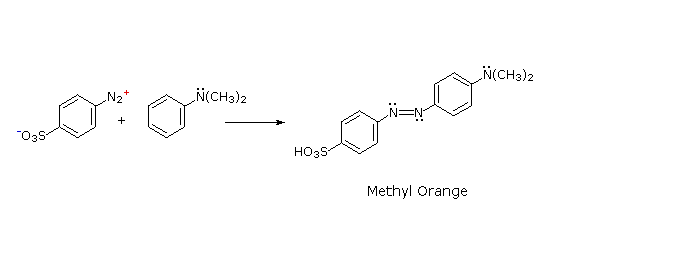
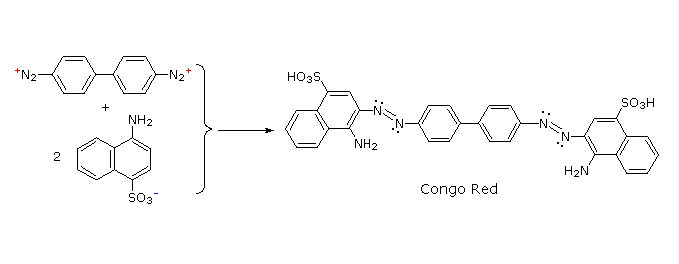
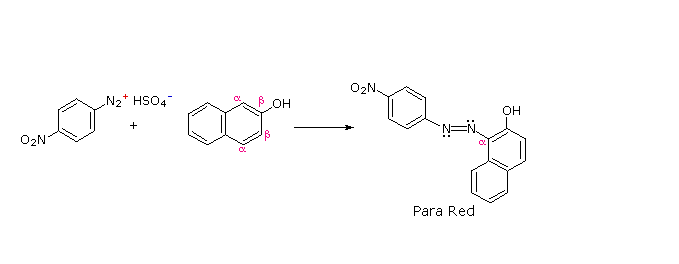
The most important application of diazo coupling reactions is electrophilic aromatic substitution of activated benzene derivatives by diazonium electrophiles.
The products of such reactions are highly colored aromatic azo compounds that find use as synthetic dyestuffs, commonly referred to as azo dyes.
Azobenzene (Y=Z=H) is light orange; however, the color of other azo compounds may range from red to deep blue depending on the nature of the aromatic rings and the substituents they carry.
Azo compounds may exist as cis/trans isomer pairs, but most of the well-characterized and stable compounds are trans.
The products of such reactions are highly colored aromatic azo compounds that find use as synthetic dyestuffs, commonly referred to as azo dyes.
Azobenzene (Y=Z=H) is light orange; however, the color of other azo compounds may range from red to deep blue depending on the nature of the aromatic rings and the substituents they carry.
Azo compounds may exist as cis/trans isomer pairs, but most of the well-characterized and stable compounds are trans.
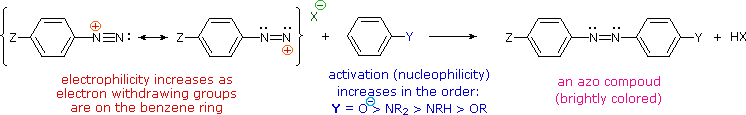
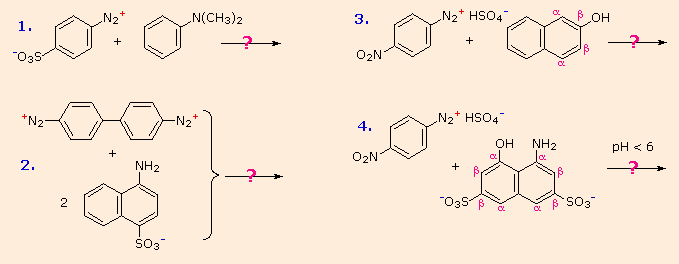
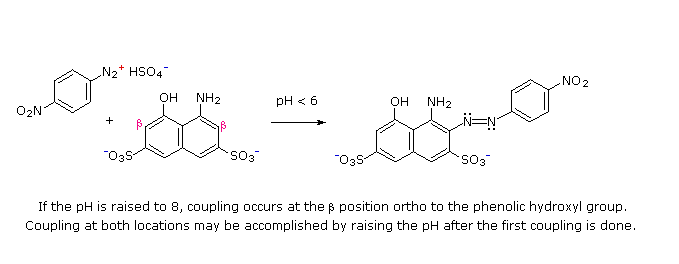
Some examples of azo coupling reactions are shown below. A few simple rules are helpful in predicting the course of such reactions:
(i) At acid pH (< 6) an amino group is a stronger activating substituent than a hydroxyl group (i.e. a phenol). At alkaline pH (> 7.5) phenolic functions are stronger activators, due to increased phenoxide base concentration.
(ii) Coupling to an activated benzene ring occurs preferentially para to the activating group if that location is free. Otherwise ortho-coupling will occur.
(iii) Naphthalene normally undergoes electrophilic substitution at an alpha-location more rapidly than at beta-sites; however, ortho-coupling is preferred. See the diagram for examples of α / β notation in naphthalenes.
You should try to conceive a plausible product structure for each of the following couplings.
answers :
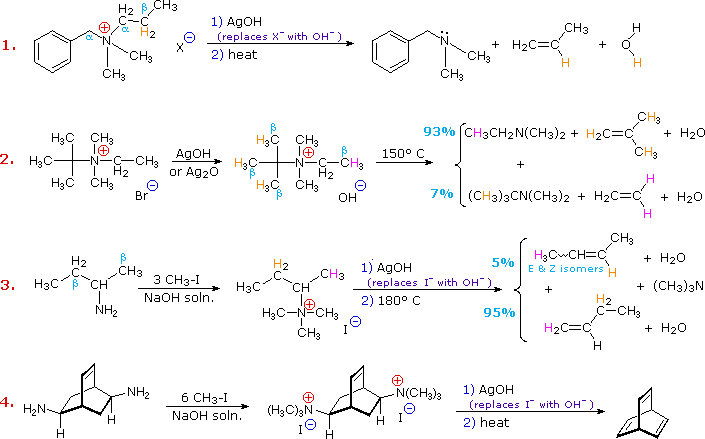
Substitution and Elimination Reactions of Amines
Substitution and Elimination Reactions of Amines
Amine functions seldom serve as leaving groups in nucleophilic substitution or base-catalyzed elimination reactions.
As noted earlier, 1º and 2º-amines are much weaker acids than alcohols, so it is not surprising that it is difficult to force the nitrogen function to assume the role of a nucleophilic leaving group. For example, heating an amine with HBr or HI does not normally convert it to the corresponding alkyl halide, as in the case of alcohols and ethers.
C6H5–N(CH3)3(+) Br(–) + R-S(–) Na(+) | acetone & heat | R-S-CH3 + C6H5–N(CH3)2 + NaBr |
(CH3)4N(+) OH(–) | heat | CH3–OH + (CH3)3N |
Hofmann Elimination
Elimination reactions of 4º-ammonium
salts are termed Hofmann eliminations.
Since the counter anion in most 4º-ammonium salts is halide, this is often
replaced by the more basic hydroxide ion through reaction with silver hydroxide
(or silver oxide).
The resulting hydroxide salt must then be heated (100 - 200 ºC) to effect the
E2-like elimination of a 3º-amine.
Example #1 below shows a typical Hofmann elimination. Obviously, for an
elimination to occur one of the alkyl substituents on nitrogen must have one or
more beta-hydrogens .
Since the counter anion in most 4º-ammonium salts is halide, this is often replaced by the more basic hydroxide ion through reaction with silver hydroxide (or silver oxide).
The resulting hydroxide salt must then be heated (100 - 200 ºC) to effect the E2-like elimination of a 3º-amine.
Example #1 below shows a typical Hofmann elimination. Obviously, for an elimination to occur one of the alkyl substituents on nitrogen must have one or more beta-hydrogens .
example ;
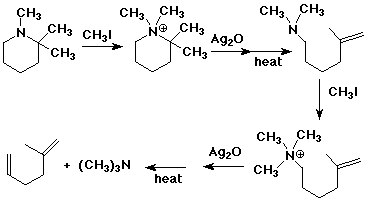
In example #2 above, two of the alkyl substituents on nitrogen
have beta-hydrogens, all of which are on methyl groups (colored orange &
magenta).
The chief product from the elimination is the alkene having the more highly substituted double bond, reflecting not only the 3:1 numerical advantage of those beta-hydrogens, but also the greater stability of the double bond.
Example #3 illustrates two important features of the Hofmann elimination:
1. Simple amines are
easily converted to the necessary 4º-ammonium salts by exhaustive alkylation,
usually with methyl iodide (methyl has no beta-hydrogens and cannot compete in
the elimination reaction). Exhaustive methylation is shown again in example #4.
2. When a given alkyl
group has two different sets of beta-hydrogens available to the elimination
process (colored orange & magenta here), the major product is often the
alkene isomer having the less substituted double bond.
The tendency of Hofmann eliminations to give the less-substituted
double bond isomer is commonly referred to as the Hofmann Rule, and contrasts strikingly with the Zaitsev
Rule formulated for dehydrohalogenations and dehydrations.
In cases where other activating groups, such as phenyl or carbonyl, are present, the Hofmann Rule may not apply. Thus, if 2-amino-1-phenylpropane is treated in the manner of example #3, the product consists largely of 1-phenylpropene (E & Z-isomers).
To understand why the base-induced elimination of 4º-ammonium salts behaves differently from that of alkyl halides it is necessary to reexamine the nature of the E2 transition state, first described for dehydrohalogenation.
The energy diagram shown earlier for a single-step bimolecular E2 mechanism is repeated below.
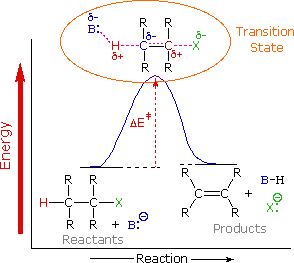
.
Three additional examples of the
Hofmann elimination are shown in the following diagram. Example #1 is
interesting in two respects.
First, it generates a 4º-ammonium halide salt in a manner different from exhaustive methylation.
Second, this salt is not converted to its hydroxide analog prior to elimination.
A concentrated aqueous solution of the halide salt is simply dropped into a refluxing sodium hydroxide solution, and the volatile hydrocarbon product is isolated by distillation
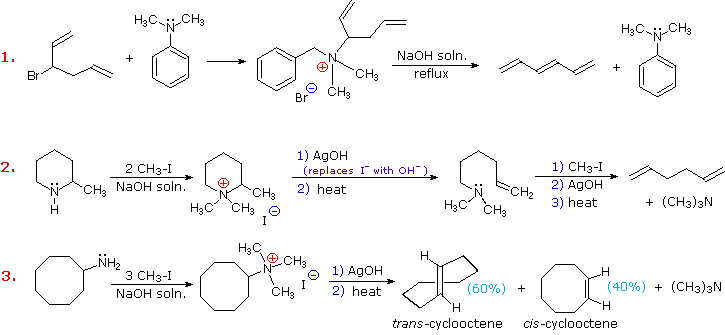
Example #2 illustrates
an important aspect of the Hofmann elimination.
If the nitrogen atom is part of a ring, then a single application of this elimination procedure does not remove the nitrogen as a separate 3º-amine product.
In order to sever the nitrogen function from the molecule, a second Hofmann elimination must be carried out.
Indeed, if the nitrogen atom was a member of two rings (fused or spiro), then three repetitions of the Hofmann elimination would be required to sever the nitrogen from the remaining molecular framework.
Example #3 is noteworthy because the less stable trans-cyclooctene is the chief product, accompanied by the cis-isomer.
An anti-E2-transition state would necessarily give the cis-cycloalkene, so the trans-isomer must be generated by a syn-elimination.
The cis-cyclooctene produced in this reaction could also be formed by a syn-elimination.
Cyclooctane is a conformationally complex structure.
Several puckered conformations that avoid angle strain are possible, and one of the most stable of these is shown on the right.
Some eclipsed bonds occur in all these conformers, and transannular hydrogen crowding is unavoidable.
Since the trimethylammonium substituent is large (about the size of tert-butyl) it will probably assume an equatorial-like orientation to avoid steric crowding.
An anti-E2 transition state is likely to require an axial-like orientation of this bulky group, making this an unfavorable path.
Cope Elimination
Cope Elimination

The Cope Reaction of N-oxides, which can easily be
prepared in situ from tertiary amines with an oxidant such as
peracid, leads to alkenes via a thermally induced syn-elimination
in aprotic solvents.
Mechanism of the Cope Elimination
The Cope Elimination is a syn periplanar elimination
in which six electrons move in a five-membered ring according to a concerted,
thermally-induced mechanism to yield an alkene and a hydroxylamine:

The
sterically demanding amine oxide function reacts preferentially with the more
easily accessible hydrogens, and often gives good selectivity favoring the
less-substituted alkene. Thus, for simple alkenes, the reaction follows the Hofmann Rule.
Cope elimination is a pyrolytic syn elimination .
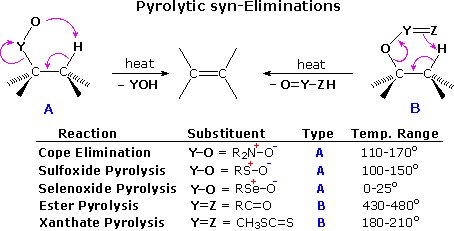
stereochemistry of reaction

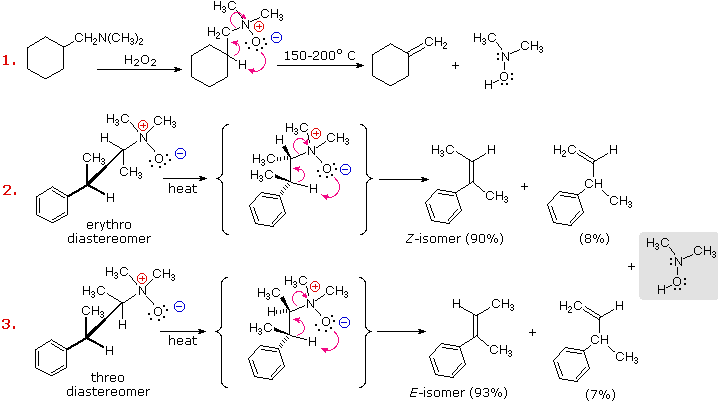
example :

A large part of Alfa Chemistry's customers are pharmaceutical and biotechnology companies, including Pfizer, Novartis, Merck & Co., Johnson & Johnson, AstraZeneca, and Bayer. Alfa Chemistry is also a preferred partner for many universities and non-profit institutes. N-butyl,methylpyrrolidinium bis((trifluoromethyl)sulfonyl)imide
ReplyDelete